Dyrektywa Komisji 2006/86/WE z dnia 24 października 2006 r. wykonująca dyrektywę 2004/23/WE Parlamentu Europejskiego i Rady w zakresie wymagań dotyczących możliwości śledzenia, powiadamiania o poważnych i niepożądanych reakcjach i zdarzeniach oraz niektórych wymagań technicznych dotyczących kodowania, przetwarzania, konserwowania, przechowywania i dystrybucji tkanek i komórek ludzkich
Dziennik Urzędowy L 294 , 25/10/2006 P. 0032 - 0050






















Dyrektywa Komisji 2006/86/WEz dnia 24 października 2006 r.wykonująca dyrektywę 2004/23/WE Parlamentu Europejskiego i Rady w zakresie wymagań dotyczących możliwości śledzenia, powiadamiania o poważnych i niepożądanych reakcjach i zdarzeniach oraz niektórych wymagań technicznych dotyczących kodowania, przetwarzania, konserwowania, przechowywania i dystrybucji tkanek i komórek ludzkichKOMISJA WSPÓLNOT EUROPEJSKICH, uwzględniając Traktat ustanawiający Wspólnotę Europejską, uwzględniając dyrektywę 2004/23/WE Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r. w sprawie ustalenia norm jakości i bezpiecznego oddawania, pobierania, testowania, przetwarzania, konserwowania, przechowywania i dystrybucji tkanek i komórek ludzkich [1], w szczególności jej art. 8, art. 11 ust. 4 oraz art. 28 lit. a), c), g) i h), a także mając na uwadze, co następuje: (1) Dyrektywa 2004/23/WE ustanawia normy jakości i bezpiecznego oddawania, pobierania, testowania, przetwarzania, konserwowania, przechowywania i dystrybucji tkanek i komórek ludzkich przeznaczonych do stosowania u ludzi oraz wytwarzanych produktów uzyskanych z tkanek i komórek ludzkich przeznaczonych do stosowania u ludzi w sposób zapewniający wysoki poziom ochrony zdrowia ludzkiego. (2) Aby zapobiec przenoszeniu się chorób poprzez tkanki i komórki ludzkie przeznaczone do stosowania u ludzi oraz aby zapewnić odpowiedni poziom jakości i bezpieczeństwa, dyrektywa 2004/23/WE wzywa do ustanowienia określonych wymagań technicznych dla każdego etapu w procesie wykorzystywania tkanek i komórek ludzkich, w tym norm i specyfikacji dotyczących systemu jakości w bankach tkanek. (3) Zgodnie z dyrektywą 2004/23/WE, aby zapewnić wysoki poziom ochrony zdrowia ludzkiego w państwach członkowskich należy ustanowić system akredytowania, mianowania, autoryzowania lub licencjonowania banków tkanek i procesów przygotowywania w bankach tkanek. Niezbędne jest ustanowienie wymagań technicznych dla takiego systemu. (4) Wymagania dotyczące systemu akredytowania, mianowania, autoryzowania lub licencjonowania banków tkanek powinny obejmować organizację i zarządzanie, personel, sprzęt i materiały, zakłady/pomieszczenia, dokumentację i rejestry oraz przegląd jakości. Akredytowane, mianowane, autoryzowane lub licencjonowane banki tkanek powinny spełniać dodatkowe wymagania dotyczące prowadzonych przez nie działalności. (5) Poziom jakości powietrza podczas przetwarzania tkanek i komórek jest kluczowym czynnikiem, który może mieć wpływ na zagrożenie zanieczyszczeniem tkanki lub komórki. Zasadniczo należy zapewnić poziom jakości powietrza o liczbie cząsteczek i liczbie kolonii drobnoustrojów odpowiadających liczbom klasy A zgodnie z "Przewodnikiem dobrej praktyki wytwarzania", załącznik 1 i dyrektywą Komisji 2003/94/WE [2]. Jednakże w niektórych sytuacjach poziom jakości powietrza o liczbie cząsteczek i liczbie kolonii drobnoustrojów odpowiadających normie klasy A nie jest określony. W takim przypadku należy przedstawić dowody poparte dokumentacją potwierdzające, że wybrane środowisko zapewnia jakość i bezpieczeństwo wymagane dla danego typu tkanki i komórki oraz dla danego procesu i zastosowania u ludzi. (6) Niniejsza dyrektywa powinna objąć swoim zakresem jakość i bezpieczeństwo tkanek i komórek ludzkich w trakcie kodowania, przetwarzania, konserwowania, przechowywania i dystrybucji do placówek opieki zdrowotnej, w których będą wszczepione do ciała ludzkiego. Dyrektywa nie powinna jednak obejmować zastosowań tych tkanek i komórek u ludzi (takich jak chirurgia implantacyjna, perfuzja, zapłodnienie lub transfer embrionów). Przepisy niniejszej dyrektywy dotyczące możliwości śledzenia i powiadamiania o poważnych i niepożądanych reakcjach i zdarzeniach stosuje się również w odniesieniu do oddawania, pobierania i testowania tkanek i komórek ludzkich podlegających dyrektywie Komisji 2006/17/WE [3]. (7) Zastosowanie tkanek i komórek u ludzi pociąga za sobą ryzyko przenoszenia chorób i wystąpienia innych niepożądanych skutków u biorców. Aby monitorować i ograniczyć takie skutki, należy określić szczególne wymagania dotyczące śledzenia oraz wspólnotową procedurę powiadamiania o poważnych i niepożądanych reakcjach i zdarzeniach. (8) Należy bezzwłocznie powiadamiać właściwy organ o podejrzeniu wystąpienia poważnych i niepożądanych reakcji u dawcy lub biorcy oraz poważnych i niepożądanych zdarzeń występujących między pobraniem a dystrybucją tkanek lub komórek, które mogą wpływać na jakość i bezpieczeństwo tkanek lub komórek i które mogą być związane z pobieraniem (w tym oceną i doborem dawcy), testowaniem, przetwarzaniem, konserwowaniem, przechowywaniem i dystrybucją tkanek i komórek ludzkich. (9) Poważne i niepożądane reakcje można wykryć w trakcie oddawania lub po oddaniu tkanek lub komórek u żywego dawcy bądź w trakcie stosowania lub po zastosowaniu u ludzi. Powinno się o nich powiadamiać partnerski bank tkanek w celu przeprowadzenia dalszego badania i zgłoszenia właściwemu organowi. Nie powinno to wykluczać możliwości bezpośredniego powiadamiania właściwego organu przez instytucje pobierające tkanki lub komórki bądź instytucje odpowiedzialne za stosowanie ich u ludzi, jeśli wyrażają one taką wolę. Niniejsza dyrektywa powinna określać minimalne dane niezbędne do powiadamiania właściwego organu, bez uszczerbku dla możliwości utrzymania lub wprowadzenia przez państwa członkowskie na swoim terytorium bardziej rygorystycznych środków ochronnych zgodnych z wymaganiami Traktatu. (10) W celu zminimalizowania kosztów przesyłania danych, uniknięcia nakładania się działań i zwiększenia skuteczności administracyjnej, do wykonania zadań związanych z przesyłaniem i przetwarzaniem informacji powinny być wykorzystane nowoczesne technologie i administracja elektroniczna. Technologie te powinny opierać się na standardowym formacie wymiany informacji przy wykorzystaniu systemu odpowiedniego dla zarządzania danymi odniesienia. (11) Aby ułatwić śledzenie i informowanie o podstawowych cechach i właściwościach tkanek i komórek, należy określić podstawowe dane, które powinny być zawarte w jednolitym kodeksie europejskim. (12) Niniejsza dyrektywa uwzględnia prawa podstawowe i przestrzega zasad ustanowionych w szczególności w Karcie praw podstawowych Unii Europejskiej. (13) Środki przewidziane w niniejszej dyrektywie są zgodne z opinią Komitetu ustanowionego zgodnie z art. 29 dyrektywy 2004/23/WE, PRZYJMUJE NINIEJSZĄ DYREKTYWĘ: Artykuł 1 Zakres 1. Niniejszą dyrektywę stosuje się do kodowania, przetwarzania, konserwowania, przechowywania i dystrybucji: a) tkanek i komórek ludzkich przeznaczonych do stosowania u ludzi; oraz b) wytwarzanych produktów uzyskanych z tkanek i komórek ludzkich przeznaczonych do stosowania u ludzi, w przypadku gdy produkty te nie są objęte innymi dyrektywami. 2. Przepisy art. 5–9 niniejszej dyrektywy dotyczące możliwości śledzenia i powiadamiania o poważnych i niepożądanych reakcjach i zdarzeniach stosuje się również w odniesieniu do oddawania, pobierania i testowania tkanek i komórek ludzkich. Artykuł 2 Definicje Do celów niniejszej dyrektywy stosuje się następujące definicje: a) "komórki reprodukcyjne" oznaczają wszystkie tkanki i komórki przeznaczone do wykorzystania do celów wspomaganego rozrodu; b) "oddawanie partnerskie" oznacza oddawanie komórek rozrodczych między mężczyzną a kobietą, których łączy intymny związek fizyczny; c) "system jakości" oznacza strukturę organizacyjną, określone obowiązki, procedury, procesy i zasoby wykorzystywane w celu wdrożenia zarządzania jakością oraz obejmuje wszystkie działania wpływające w sposób pośredni lub bezpośredni na jakość; d) "zarządzanie jakością" oznacza skoordynowane działania mające na celu kierowanie i kontrolowanie instytucji w zakresie jakości; e) "standardowe procedury operacyjne (SPO)" oznaczają pisemne instrukcje opisujące etapy określonego procesu, w tym wykorzystywane materiały i metody oraz oczekiwany produkt końcowy; f) "walidacja" (lub "kwalifikacja" w przypadku sprzętu lub środowiska) oznacza sporządzenie udokumentowanych dowodów dających wysoki stopień pewności, że określony proces, sprzęt lub otoczenie pozwolą na stałe uzyskiwanie produktu zgodnego z uprzednio ustalonymi specyfikacjami i właściwościami pod względem jakości; proces jest zatwierdzany pod kątem wydajności systemu w odniesieniu do jego skuteczności w oparciu o zamierzone użycie; g) "możliwość śledzenia" oznacza zdolność lokalizowania i identyfikowania tkanki/komórki na dowolnym etapie od jej pobrania, w trakcie przetwarzania, testowania i przechowywania, do jej dystrybucji do biorcy lub utylizacji, pociągająca za sobą zdolność zidentyfikowania dawcy i banku tkanek lub zakładu wytwórczego odbierającego, przetwarzającego lub przechowującego tkankę/komórki oraz zdolność zidentyfikowania biorcy/biorców w placówce/placówkach medycznych stosującej(-ych) tkankę/komórki u biorcy; możliwość śledzenia dotyczy również zdolności lokalizowania i identyfikowania wszelkich istotnych danych związanych z produktami oraz materiałami mającymi kontakt z tkankami/komórkami; h) "krytyczny" oznacza mający potencjalny wpływ na jakość i/lub bezpieczeństwo bądź będący w kontakcie z komórkami i tkankami; i) "organizacja pobierająca" oznacza placówkę medyczną lub oddział szpitala lub inną jednostkę, która prowadzi działania polegające na pobieraniu tkanek i komórek ludzkich i może nie być akredytowana, mianowana, autoryzowana lub licencjonowana jako bank tkanek; j) "organizacja odpowiedzialna za stosowanie u ludzi" oznacza placówkę medyczną lub oddział szpitala lub inną jednostkę, która stosuje tkanki i komórki ludzkie u ludzi. Artykuł 3 Wymagania dotyczące akredytowania, mianowania, autoryzowania lub licencjonowania banków tkanek Bank tkanek musi spełniać wymagania określone w załączniku I. Artykuł 4 Wymagania dotyczące akredytowania, mianowania, autoryzowania lub licencjonowania procesów przygotowywania tkanek i komórek Procesy przygotowywania w bankach tkanek muszą spełniać wymagania określone w załączniku II. Artykuł 5 Powiadamianie o poważnych i niepożądanych reakcjach 1. Państwa członkowskie dbają o to, aby: a) organizacje pobierające wprowadziły procedury zachowywania rejestrów pobranych tkanek i komórek oraz bezzwłocznego powiadamiania banków tkanek o wszelkich poważnych i niepożądanych reakcjach u żywych dawców, które mogą wpływać na jakość i bezpieczeństwo tkanek lub komórek; b) organizacje odpowiedzialne za stosowanie tkanek i komórek u ludzi wprowadziły procedury zachowywania rejestrów wykorzystanych tkanek i komórek oraz procedury bezzwłocznego powiadamiania banków tkanek o wszelkich poważnych i niepożądanych reakcjach zaobserwowanych w trakcie lub po klinicznym zastosowaniu, które mogą być związane z jakością i bezpieczeństwem tkanek i komórek; c) banki tkanek, które wydają tkanki i komórki do stosowania u ludzi, przedstawiły organizacji odpowiedzialnej za stosowanie tkanek i komórek u ludzi informacje na temat sposobu, w jaki organizacja ta powinna zgłaszać poważne i niepożądane reakcje, o których mowa w lit. b). 2. Państwa członkowskie dbają o to, aby banki tkanek: a) wprowadziły procedury bezzwłocznego przekazywania właściwemu organowi wszystkich właściwych i dostępnych informacji na temat podejrzanych, poważnych i niepożądanych reakcji, o których mowa w ust. 1 lit. a) i b); b) wprowadziły procedury bezzwłocznego przekazywania właściwemu organowi informacji na temat wniosków z badania mającego na celu przeanalizowanie przyczyn i konsekwencji. 3. Państwa członkowskie dbają o to, aby: a) osoba odpowiedzialna, o której mowa w art. 17 dyrektywy 2004/23/WE, zgłaszała właściwemu organowi informacje zawarte w powiadomieniu określonym w załączniku III część A; b) banki tkanek powiadamiały właściwy organ o działaniach podejmowanych w odniesieniu do innych tkanek i komórek, których mogą dotyczyć poważne i niepożądane reakcje, które zostały wydane w celu zastosowania u ludzi; c) banki tkanek powiadamiały właściwy organ o wnioskach z badania, przedstawiając co najmniej informacje określone w załączniku III część B. Artykuł 6 Powiadamianie o poważnych i niepożądanych zdarzeniach 1. Państwa członkowskie dbają o to, aby: a) organizacje pobierające i banki tkanek wprowadziły procedury zachowywania rejestrów i bezzwłocznego powiadamiania banków tkanek o wszelkich poważnych i niepożądanych zdarzeniach, które następują w trakcie pobierania i które mogą wpływać na jakość i/lub bezpieczeństwo tkanek lub komórek; b) organizacje odpowiedzialne za stosowanie tkanek i komórek u ludzi wprowadziły procedury bezzwłocznego powiadamiania banków tkanek o wszelkich poważnych i niepożądanych zdarzeniach, które mogą wpływać na jakość i bezpieczeństwo tkanek i komórek; c) banki tkanek przedstawiły organizacji odpowiedzialnej za stosowanie tkanek i komórek u ludzi informacje na temat sposobu, w jaki organizacja ta powinna zgłaszać im poważne i niepożądane zdarzenia, które mogą wpływać na jakość i bezpieczeństwo tkanek i komórek. 2. W przypadku wspomaganego rozrodu, każdego rodzaju błędną identyfikację lub pomylenie komórek rozrodczych, gamet lub embrionów uważa się za poważne i niepożądane zdarzenie. Wszystkie osoby, organizacje pobierające lub organizacje odpowiedzialne za stosowanie tkanek i komórek u ludzi, przeprowadzające wspomagany rozród, zgłaszają takie zdarzenia bankom dostarczającym tkanki w celu przeprowadzenia badania i powiadomienia właściwego organu. 3. Państwa członkowskie dbają o to, aby banki tkanek: a) wprowadziły procedury bezzwłocznego przekazywania właściwemu organowi wszystkich właściwych i dostępnych informacji na temat podejrzanych, poważnych i niepożądanych reakcji, o których mowa w ust. 1 lit. a) i b); b) wprowadziły procedury bezzwłocznego informowania właściwego organu o wnioskach z badania mającego na celu przeanalizowanie przyczyn i konsekwencji. 4. Państwa członkowskie dbają o to, aby: a) osoba odpowiedzialna, o której mowa w art. 17 dyrektywy 2004/23/WE, powiadamiała właściwy organ o informacjach zawartych w powiadomieniu określonym w załączniku IV część A; b) banki tkanek oceniały poważne i niepożądane zdarzenia w celu określenia przyczyn w trakcie procesu, których można uniknąć; c) banki tkanek powiadamiały właściwy organ o wnioskach z badania, przedstawiając co najmniej informacje określone w załączniku IV część B. Artykuł 7 Sprawozdania roczne 1. Do dnia 30 czerwca następnego roku państwa członkowskie przedstawiają Komisji sprawozdanie roczne dotyczące powiadamiania o poważnych i niepożądanych reakcjach i zdarzeniach przekazane właściwemu organowi. Komisja przekazuje właściwym organom państw członkowskich streszczenie otrzymanych sprawozdań. Właściwy organ udostępnia takie sprawozdanie bankom tkanek. 2. Przesyłanie danych opiera się na standardowym formacie wymiany informacji określonym w załączniku V, części A i B, i obejmuje wszystkie informacje niezbędne do zidentyfikowania nadawcy i zachowanie danych odniesienia dotyczących nadawcy. Artykuł 8 Wymiana informacji między właściwymi organami a Komisją Państwa członkowskie dbają o to, aby właściwe organy przekazywały sobie nawzajem i Komisji właściwe informacje na temat poważnych i niepożądanych reakcji i zdarzeń w celu zagwarantowania podjęcia odpowiednich działań. Artykuł 9 Możliwość śledzenia 1. Banki tkanek posiadają precyzyjny i skuteczny system jednoznacznego identyfikowania i znakowania otrzymywanych i wydawanych komórek/tkanek. 2. Banki tkanek i organizacje odpowiedzialne za stosowanie tkanek i komórek u ludzi zachowują dane określone w załączniku VI przez okres co najmniej trzydziestu lat, we właściwej formie nadającej się do odczytania. Artykuł 10 Europejski system kodowania 1. Aby zapewnić właściwą identyfikację dawcy i możliwość śledzenia wszystkich pobranych materiałów oraz przekazywanie informacji dotyczących podstawowych cech i właściwości tkanek i komórek, każdemu materiałowi pobranemu w banku tkanek jest przydzielany jednolity europejski kod identyfikacyjny. Kod zawiera co najmniej informacje określone w załączniku VII. 2. Ustępu 1 nie stosuje się do oddawania partnerskiego komórek rozrodczych. Artykuł 11 Transpozycja 1. Państwa członkowskie wprowadzają w życie przepisy ustawowe, wykonawcze i administracyjne niezbędne do wykonania niniejszej dyrektywy najpóźniej do dnia 1 września 2007 r. Państwa członkowskie niezwłocznie przekazują Komisji tekst tych przepisów oraz tabelę korelacji między tymi przepisami a niniejszą dyrektywą. Państwa członkowskie wprowadzają w życie przepisy ustawowe, wykonawcze i administracyjne niezbędne do wykonania art. 10 niniejszej dyrektywy do dnia 1 września 2008 r. Przepisy przyjęte przez państwa członkowskie zawierają odesłanie do niniejszej dyrektywy lub odesłanie takie towarzyszy ich urzędowej publikacji. Metody dokonania takiego odesłania określane są przez państwa członkowskie. 2. Państwa członkowskie przedstawiają Komisji teksty najważniejszych przepisów prawa krajowego, które przyjmują w dziedzinie objętej niniejszą dyrektywą. Artykuł 12 Wejście w życie Niniejsza dyrektywa wchodzi w życie dwudziestego dnia po jej opublikowaniu w Dzienniku Urzędowym Unii Europejskiej. Artykuł 13 Adresaci Niniejsza dyrektywa skierowana jest do państw członkowskich. Sporządzono w Brukseli, dnia 24 października 2006 r. W imieniu Komisji Markos Kyprianou Członek Komisji [1] Dz.U. L 102 z 7.4.2004, str. 48. [2] http://pharmacos.eudra.org/F2/eudralex/vol-4/home.htm oraz Dz.U. L 262 z 14.10.2003, str. 22. [3] Dz.U. L 38 z 9.2.2006, str. 40.
ZAŁĄCZNIK I Wymagania dotyczące akredytowania, mianowania, autoryzowania lub licencjonowania banków tkanek, o których mowa w art. 3 A. ORGANIZACJA I ZARZĄDZANIE 1. Należy wyznaczyć osobę odpowiedzialną, posiadającą kwalifikacje i pełniącą obowiązki przewidziane w art. 17 dyrektywy 2004/23/WE. 2. Bank tkanek musi posiadać strukturę organizacyjną i procedury operacyjne odpowiednie dla działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, musi istnieć schemat organizacyjny, w którym jasno określone są uprawnienia i odpowiedzialność. 3. Każdy bank tkanek musi mieć dostęp do zarejestrowanego lekarza medycyny wyznaczonego na doradcę i sprawującego nadzór nad prowadzonymi przez placówki działalnościami medycznymi, takimi jak dobór dawcy, przegląd wyników klinicznych stosowanych tkanek i komórek lub, jeśli to stosowne, współdziałania z użytkownikami klinicznymi. 4. W odniesieniu do działalności, które mają zostać akredytowane/mianowane/autoryzowane lub licencjonowane, musi być stosowany udokumentowany system zarządzania jakością zgodny z normami ustanowionymi w niniejszej dyrektywie. 5. Należy zapewnić identyfikację i ograniczenie w możliwie największym stopniu ryzyka, które jest nieodłącznym elementem stosowania i obchodzenia się z materiałem biologicznym, przy jednoczesnym zachowaniu jakości i bezpieczeństwa odpowiednich dla przeznaczenia tkanek i komórek. Ryzyko obejmuje charakterystyczne dla banku tkanek ryzyko związane w szczególności z procedurami, otoczeniem i stanem zdrowia personelu. 6. Umowy zawierane między bankami tkanek a stronami trzecimi muszą być zgodne z art. 24 dyrektywy 2004/34/WE. Umowy zawierane ze stronami trzecimi muszą określać zarówno warunki relacji i obowiązków, jak i protokoły niezbędne do spełnienia wymaganych specyfikacji pracy. 7. Należy wprowadzić udokumentowany system nadzorowany przez osobę odpowiedzialną, dzięki któremu można potwierdzić, że tkanki i/lub komórki spełniają właściwe wymagania specyfikacji w zakresie bezpieczeństwa i jakości, pozwalające na ich dopuszczenie i dystrybucję. 8. W przypadku zakończenia działalności zawarte umowy i procedury przyjęte zgodnie z art. 21 ust. 5 dyrektywy 2004/23/WE obejmują dane dotyczące śledzenia i materiały dotyczące jakości i bezpieczeństwa komórek i tkanek. 9. Należy wprowadzić udokumentowany system zapewniający identyfikację każdej jednostki tkanki lub komórek na każdym etapie działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane. B. PERSONEL 1. W bankach tkanek należy zapewnić wystarczającą liczbę personelu posiadającego kwalifikacje do wykonywanych zadań. Kompetencje personelu muszą być poddawane ocenie we właściwych odstępach czasu określonych w systemie jakości. 2. Każdy pracownik powinien mieć jasny, udokumentowany i aktualny opis zakresu swoich obowiązków. Zadania, obowiązki i odpowiedzialność pracowników muszą być jasno udokumentowane i zrozumiałe. 3. Personelowi należy zapewnić szkolenia wstępne/podstawowe, szkolenia uaktualniane, jeśli wymagają tego zmiany procedur lub rozwój wiedzy naukowej, oraz odpowiednie możliwości właściwego rozwoju zawodowego. Program szkolenia musi zapewnić, popierając odpowiednimi dokumentami, aby każda osoba: a) wykazała umiejętności w realizowaniu wyznaczonych zadań; b) posiadała odpowiednią wiedzę i rozumiała procesy naukowe/techniczne i zasady dotyczące wyznaczonych zadań; c) rozumiała strukturę organizacyjną, system jakości i zasady zdrowia i bezpieczeństwa placówki, w której pracuje; oraz d) była odpowiednio poinformowana o etycznych, prawnych i regulacyjnych aspektach wykonywanej pracy. C. SPRZĘT I MATERIAŁY 1. Cały sprzęt i wszystkie materiały muszą być zaprojektowane i utrzymywane, tak aby odpowiadały swojemu przeznaczeniu oraz powinny ograniczać w możliwie największym stopniu jakiekolwiek zagrożenie dla biorców i/lub personelu. 2. Cały sprzęt krytyczny i wszystkie krytyczne urządzenia techniczne muszą być zidentyfikowane i zatwierdzone, poddawane regularnym kontrolom i profilaktycznej konserwacji zgodnie z instrukcjami producenta. W przypadku gdy sprzęt i materiały dotyczą krytycznych parametrów przetwarzania lub przechowywania (np. temperatura, ciśnienie, liczba cząsteczek, poziomy zakażenia drobnoustrojami), muszą one być jako takie zidentyfikowane i, w stosownych przypadkach, poddane odpowiedniemu monitorowaniu, powiadamianiu, alarmowaniu i działaniu naprawczemu w celu wykrycia nieprawidłowego funkcjonowania i wad oraz w celu zapewnienia stałego utrzymywania krytycznych parametrów w dopuszczalnych granicach. Cały sprzęt posiadający krytyczne funkcje pomiarowe musi być skalibrowany zgodnie z możliwą do ustalenia normą, jeśli taka istnieje. 3. Nowy i naprawiany sprzęt musi być przetestowany po instalacji i zatwierdzony przed użyciem. Wyniki testu należy udokumentować. 4. Konserwacja, przeglądy, czyszczenie, dezynfekcja i asenizacja całego sprzętu krytycznego muszą być wykonywane regularnie i rejestrowane w odpowiedni sposób. 5. Muszą być dostępne procedury dotyczące działania każdego elementu sprzętu krytycznego opisujące w szczegółowy sposób działania, które należy podjąć w przypadku nieprawidłowego funkcjonowania lub awarii. 6. Procedury dotyczące działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, muszą w szczegółowy sposób opisywać specyfikacje wszystkich krytycznych materiałów i odczynników. W szczególności muszą być określone specyfikacje dotyczące dodatków (np. roztworów) i materiałów opakowaniowych. Odczynniki i materiały krytyczne muszą być zgodne z udokumentowanymi wymaganiami i specyfikacjami oraz w stosownych przypadkach z wymaganiami dyrektywy Rady 93/42/EWG z dnia 14 czerwca 1993 r. dotyczącej wyrobów medycznych [1] i dyrektywy 98/79/WE Parlamentu Europejskiego i Rady z dnia 27 października 1998 r. w sprawie wyrobów medycznych używanych do diagnozy in vitro [2]. D. OBIEKTY/POMIESZCZENIA 1. Aby prowadzić działalności, które mają zostać akredytowane/mianowane/autoryzowane lub licencjonowane, bank tkanek musi dysponować odpowiednimí obiektami zgodnymi z normami ustanowionymi w niniejszej dyrektywie. 2. Jeśli działalności te obejmują przetwarzanie tkanek i komórek przy wystawieniu na działanie otoczenia, musi ono być prowadzone w otoczeniu o określonej jakości powietrza i czystości w celu zminimalizowania ryzyka zakażenia, w tym zakażenia krzyżowego między dawcami. Skuteczność tych środków musi być zatwierdzona i monitorowana. 3. O ile w pkt 4 nie wskazano inaczej, w przypadku gdy tkanki lub komórki są wystawione na działanie otoczenia w trakcie przetwarzania, bez późniejszego procesu inaktywacji drobnoustrojów, wymagana jest jakość powietrza o liczbie cząsteczek i liczbie kolonii drobnoustrojów odpowiadających liczbom klasy A zgodnie z obowiązującym obecnie "Przewodnikiem dobrej praktyki wytwarzania", załącznik 1 i dyrektywą 2003/9/WE, przy czym tło otoczenia musi być dostosowane do procesu przetwarzania danej tkanki/komórki, a w zakresie liczby cząsteczek i liczby drobnoustrojów co najmniej odpowiadać klasie D dobrej praktyki wytwarzania. 4. Otoczenie mniej rygorystyczne niż określone w pkt 3 dopuszcza się w przypadku gdy: a) stosuje się zatwierdzony proces inaktywacji drobnoustrojów lub zatwierdzony proces sterylizacji końcowej; lub b) wykazano, że wystawienie na działanie otoczenia klasy A ma negatywny wpływ na wymagane własności danej tkanki lub komórki; lub c) wykazano, że sposób i tryb stosowania tkanki lub komórki u biorcy pociągają za sobą znacznie niższe ryzyko przenoszenia infekcji bakteryjnych lub grzybicy na biorcę niż w przypadku transplantacji komórki i tkanki; lub d) przeprowadzenie wymaganego procesu w otoczeniu klasy A (np. ze względu na wymagania dotyczące określonego sprzętu w strefie przetwarzania, który nie odpowiada w pełni klasie A), jest technicznie niemożliwe. 5. W sytuacjach opisanych w pkt 4 lit. a)–d) należy określić otoczenie. Należy wykazać, popierając dokumentacją, że wybrane otoczenie spełnia wymagania dotyczące jakości i bezpieczeństwa, uwzględniając co najmniej zamierzone przeznaczenie, sposób stosowania i stan odporności biorcy. W każdym właściwym dziale banku tkanek należy zapewnić zarówno odpowiednie ubrania i sprzęt przeznaczone do ochrony osobistej i higieny, jak i pisemne instrukcje dotyczące higieny i ubioru. 6. W przypadku gdy działalności, które mają zostać akredytowane/mianowane/autoryzowane lub licencjonowane, wiążą się z przechowywaniem tkanek i komórek, należy określić warunki przechowywania niezbędne do zachowania wymaganych właściwości tkanki i komórki, w tym właściwe parametry takie jak temperatura, wilgotność lub jakość powietrza. 7. Parametry krytyczne (np. temperatura, wilgotność, jakość powietrza) muszą być kontrolowane, monitorowane i rejestrowane w celu wykazania ich zgodności z określonymi warunkami przechowywania. 8. Aby zapobiec wszelkim przypadkom pomylenia i zakażeniu krzyżowemu pomiędzy tkankami/komórkami, w obiektach do przechowywania należy zapewnić możliwość wyraźnego oddzielenia i rozróżnienia tkanek i komórek przed dopuszczeniem/w trakcie kwarantanny od tych, które zostały dopuszczone i od tych, które zostały odrzucone. W miejscach do przechowywania zarówno tkanek i komórek dopuszczonych, jak i tych będących w trakcie kwarantanny należy przeznaczyć fizycznie oddzielone strefy lub urządzenia do przechowywania bądź bezpieczne odizolowanie wewnątrz urządzenia, aby móc przechować tkanki i komórki zgromadzone zgodnie ze specjalnymi kryteriami. 9. Bank tkanek musi posiadać pisemne zasady i procedury dotyczące kontroli dostępu, czyszczenia i konserwacji, usuwania odpadów i reorganizacji usług w sytuacjach nadzwyczajnych. E. DOKUMENTACJA I REJESTRY 1. W odniesieniu do działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, należy wprowadzić system zapewniający jasno określone i skuteczne zasady dokumentacji, prawidłową rejestrację i rejestry oraz standardowe procedury operacyjne (SPO). Dokumenty muszą być poddawane regularnym przeglądom i spełniać normy ustanowione w niniejszej dyrektywie. System musi zapewniać standardowe wykonanie prac oraz możliwość prześledzenia wszystkich etapów, czyli kodowania, warunków kwalifikacji dawców, pobierania, testowania, przetwarzania, konserwowania, przechowywania, dystrybucji lub usuwania, w tym aspektów związanych z kontrolą jakości i zapewnieniem jakości. 2. W przypadku każdej krytycznej działalności należy określić i udokumentować potrzebne materiały, sprzęt i personel. 3. W bankach tkanek wszystkie zmiany w dokumentach muszą być kontrolowane, opatrzone datą, zatwierdzane, dokumentowane i niezwłocznie wprowadzane przez upoważniony personel. 4. Aby móc odtworzyć historię prowadzonych przeglądów dokumentów i zmian oraz aby zapewnić stosowanie jedynie obowiązujących wersji dokumentów, należy wprowadzić procedurę kontroli dokumentów. 5. Należy wykazać, że rejestry są wiarygodne i że zawierają wiarygodne wyniki. 6. Rejestry muszą być czytelne i sporządzone na trwałym nośniku, mogą być sporządzone odręcznie lub przesłane do innego zatwierdzonego systemu, jak system komputerowy bądź mikrofilm. 7. Nie naruszając przepisów art. 9 ust. 2, wszystkie rejestry, w tym dane krytyczne, niezbędne dla zapewnienia bezpieczeństwa i jakości tkanek i komórek, zachowuje się w taki sposób, aby dostęp do tych danych był zapewniony przez co najmniej dziesięć lat po upływie terminu, zastosowaniu klinicznym lub usunięciu. 8. Rejestry muszą spełniać wymagania dotyczące poufności ustanowione w art. 14 dyrektywy 2004/23/WE. Dostęp do rejestrów i danych musi być ograniczony do osób upoważnionych przez osobę odpowiedzialną i do właściwego organu do celów inspekcji i kontroli. F. PRZEGLĄD JAKOŚCI 1. W odniesieniu do działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, musi być wprowadzony system audytu. Audyt musi być przeprowadzany w sposób niezależny przez przeszkolone i kompetentne osoby, co najmniej co dwa lata, w celu sprawdzenia zgodności z zatwierdzonymi protokołami i wymogami prawnymi. Wyniki i działania naprawcze muszą być udokumentowane. 2. Przypadki nieprzestrzegania wymaganych norm jakości i bezpieczeństwa muszą być przedmiotem udokumentowanego badania, które obejmuje decyzję o możliwych działaniach naprawczych i zapobiegawczych. Decyzję o losie tkanek i komórek niezgodnych z normami należy podjąć zgodnie z procedurami pisemnymi nadzorowanymi przez osobę odpowiedzialną i następnie dokonać wpisu w rejestrze. Wszystkie tkanki i komórki niezgodne z normami muszą być zidentyfikowane i zaksięgowane. 3. Działania naprawcze należy udokumentować, rozpocząć i zakończyć we właściwym terminie i w skuteczny sposób. Po zrealizowaniu działania zapobiegawcze i naprawcze powinny zostać ocenione pod względem ich skuteczności. 4. Aby zapewnić ciągłą i systematyczną poprawę, bank tkanek powinien wprowadzić procesy umożliwiające przegląd działania systemu zarządzania jakością. [1] Dz.U. L 169 z 12.7.1993, str. 1. Dyrektywa ostatnio zmieniona rozporządzeniem (WE) nr 1882/2003 Parlamentu Europejskiego i Rady (Dz.U. L 284 z 31.10.2003, str. 1). [2] Dz.U. L 331 z 7.12.1998, str. 1. Dyrektywa zmieniona rozporządzeniem (WE) nr 1882/2003.
ZAŁĄCZNIK II Wymagania dotyczące autoryzowania procesów przygotowywania tkanek i komórek w bankach tkanek, o których mowa w art. 4 Właściwy organ autoryzuje każdy proces przygotowywania tkanki i komórki po przeprowadzeniu oceny kryteriów doboru dawcy i procedur pobierania, protokołów dla każdego etapu procesu, kryteriów zarządzania jakością oraz ostatecznych kryteriów ilościowych i jakościowych dotyczących komórek i tkanek. Ocena ta musi spełniać co najmniej wymagania określone w niniejszym Załączniku. A. ODBIÓR W BANKU TKANEK W chwili odbioru pobranych tkanek i komórek w banku tkanek, tkanki i komórki muszą spełniać wymagania określone w dyrektywie 2006/17/WE. B. PRZETWARZANIE W przypadku gdy działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, obejmują przetwarzanie tkanek i komórek, procedury w banku tkanek muszą spełniać następujące kryteria: 1) krytyczne procedury przetwarzania muszą być zatwierdzone i nie mogą sprawiać, że tkanki lub komórki są klinicznie nieskuteczne lub szkodliwe dla biorcy. Walidacja ta może być przeprowadzona w oparciu o badania wykonane przez sam bank tkanek lub dane pochodzące z opublikowanych prac bądź w przypadku procedur istniejących od wielu lat w oparciu o ocenę retrospektywną wyników klinicznych dotyczących tkanek dostarczonych przez bank; 2) należy wykazać, że zatwierdzony proces może być stale i skutecznie przeprowadzany przez personel, w otoczeniu zapewnionym przez bank tkanek; 3) procedury muszą być udokumentowane w SPO, które muszą być zgodne z zatwierdzoną metodą i normami ustanowionymi w niniejszej dyrektywie, zgodnie z załącznikiem I pkt E ppkt 1–4; 4) należy dbać o to, aby wszystkie procesy były przeprowadzane zgodnie z zatwierdzonymi SPO; 5) w przypadku gdy w odniesieniu do tkanki lub komórek stosuje się procedurę inaktywacji drobnoustrojów, należy ją określić, udokumentować i zatwierdzić; 6) przed wprowadzeniem jakiejkolwiek znaczącej zmiany w procesie przetwarzania, zmieniony proces należy zatwierdzić i udokumentować; 7) procedury przetwarzania muszą być regularnie poddawane krytycznej ocenie w celu zapewnienia osiągania wyznaczonych celów; 8) procedury usuwania tkanki i komórek muszą uniemożliwiać zakażenie innych oddanych tkanek lub komórek i produktów, otoczenia procesu przetwarzania lub personelu. Procedury te muszą spełniać wymogi przepisów krajowych. C. PRZECHOWYWANIE I DOPUSZCZANIE PRODUKTÓW W przypadku gdy działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, obejmują przechowywanie i dopuszczanie tkanek i komórek, zatwierdzone procedury w banku tkanek muszą spełniać następujące kryteria: 1) dla każdego typu warunków przechowywania musi być określony maksymalny czas przechowywania. Wybrany okres musi uwzględniać, między innymi, możliwe pogorszenie się właściwości tkanki lub komórki; 2) aby zapewnić, by tkanki i/lub komórki były dopuszczane pod warunkiem spełnienia wymagań ustanowionych w niniejszej dyrektywie, niezbędny jest system inwentaryzacji tkanek i/lub komórek. Musi istnieć standardowa procedura operacyjna, która w szczegółowy sposób opisuje okoliczności, obowiązki i procedury dopuszczania tkanek i komórek do dystrybucji; 3) system identyfikacji tkanek i komórek musi na każdym etapie przetwarzania w banku tkanek wyraźnie rozróżniać produkty dopuszczone od produktów niedopuszczonych (poddanych kwarantannie) i odrzuconych; 4) rejestry muszą wykazać, że tkanki i komórki spełniają wszystkie wymagane specyfikacje, zanim zostaną dopuszczone, w szczególności że wszystkie obowiązujące formularze zgłoszeń, właściwe dokumentacje medyczne, rejestry przetwarzania i wyniki badań zostały sprawdzone zgodnie z procedurą pisemną przez osobę upoważnioną do tego zadania przez osobę odpowiedzialną, określoną w art. 17 dyrektywy 2004/23/WE. Jeżeli do celu ogłoszenia wyników laboratoryjnych użyty jest komputer, ślad rewizyjny powinien umożliwić wskazanie osoby odpowiedzialnej za ich ogłoszenie; 5) po wprowadzeniu każdego nowego kryterium doboru dawcy lub kryterium testowania bądź każdej znaczącej zmiany dotyczącej jakiegokolwiek etapu przetwarzania, która zwiększa bezpieczeństwo lub jakość, należy przeprowadzić udokumentowaną ocenę ryzyka zatwierdzoną przez osobę odpowiedzialną, określoną w art. 17 dyrektywy 2004/23/WE, w celu określenia losu wszystkich przechowywanych tkanek i komórek. D. DYSTRYBUCJA I WYCOFANIE W przypadku gdy działalności, które mają zostać akredytowane/mianowane/autoryzowane/licencjonowane, obejmują dystrybucję tkanek i komórek, zatwierdzone procedury w banku tkanek muszą spełniać następujące kryteria: 1) muszą być określone krytyczne warunki transportu, takie jak temperatura i maksymalny termin, tak aby zachować wymagane właściwości tkanki i komórki; 2) pojemnik/paczka musi być zabezpieczona i zapewniać utrzymanie tkanki i komórek w określonych warunkach. Wszystkie pojemniki i paczki muszą być zatwierdzone jako odpowiednie do określonego celu; 3) w przypadku gdy dystrybucja jest na mocy umowy zlecona stronie trzeciej, należy sporządzić pisemne zobowiązanie gwarantujące zachowanie wymaganych warunków; 4) w banku tkanek musi być zatrudniona osoba dokonująca oceny konieczności wycofania oraz podejmująca i koordynująca niezbędne działania; 5) należy wprowadzić skuteczną procedurę wycofania obejmującą opis obowiązków i działań, które należy podjąć. Działania te obejmują powiadomienie właściwego organu; 6) działania muszą być podjęte w ciągu ustalonych wcześniej okresów i muszą obejmować prześledzenie wszystkich właściwych tkanek i komórek, a w stosownych przypadkach prześledzenie wstecz. Celem badań jest identyfikacja wszystkich dawców, którzy mogli się przyczynić do reakcji biorcy i wycofania dostępnych tkanek i komórek od tego dawcy, jak również powiadomienie odbiorców i biorców tkanek i komórek pobranych od tego samego dawcy, w przypadku gdyby byli narażeni na ryzyko; 7) należy wprowadzić procedury rozpatrywania wniosków o przyznanie tkanek i komórek. Zasady przyznawania tkanek i komórek niektórym pacjentom lub zakładom opieki zdrowotnej muszą być udokumentowane i udostępnione tym osobom lub zakładom na ich żądanie; 8) należy wprowadzić udokumentowany system obchodzenia się ze zwróconymi produktami, w tym kryteria, na podstawie których produkty te są w stosownych przypadkach zatwierdzane do wpisania w inwentarzu. E. OZNAKOWANIE KOŃCOWE DO DYSTRYBUCJI 1. Na pierwotnym pojemniku zawierającym tkankę/komórki muszą znajdować się następujące informacje: a) rodzaj tkanek i komórek, numer identyfikacyjny lub kod tkanki/komórek oraz, w stosownych przypadkach, numer serii i partii; b) dane identyfikacyjne dotyczące banku tkanek; c) data ważności; d) w przypadku oddawania autologicznego należy zaznaczyć ("tylko do użytku autologicznego") oraz określić dawcę/biorcę; e) w przypadku oddawania tkanek/komórek dla konkretnego biorcy oznaczenie musi określać zamierzonego biorcę; f) jeżeli wiadomo, że tkanki i komórki uzyskały wynik pozytywny w badaniu na obecność markera określonej choroby zakaźnej, należy oznaczyć je informacją: "ZAGROŻENIE BIOLOGICZNE". Jeżeli którakolwiek z informacji wymienionych wyżej w lit. d) i e) nie może zostać umieszczona na oznakowaniu pierwotnego pojemnika, musi ona zostać podana w oddzielnym dokumencie towarzyszącym pierwotnemu pojemnikowi. Dokument ten musi być zapakowany z pojemnikiem pierwotnym, tak aby był cały czas razem z tym pojemnikiem. 2. Na oznaczeniu lub w dokumentach towarzyszących należy umieścić następujące informacje: a) opis (definicja) oraz, w stosownych przypadkach, wymiary produktu tkanki lub komórki; b) w stosownych przypadkach, morfologia i dane dotyczące funkcjonowania; c) data dystrybucji tkanki/komórek; d) dawki biologiczne zastosowane u dawcy oraz wyniki; e) zalecenia dotyczące przechowywania; f) instrukcje dotyczące otwierania pojemnika, paczki i wszelkich niezbędnych czynności/przywrócenia do stanu poprzedniego; g) data ważności po otwarciu/wykonaniu innych czynności; h) instrukcje dotyczące powiadamiania o poważnych i niepożądanych reakcjach i/lub zdarzeniach zgodnie z art. 5–6; i) obecność potencjalnie szkodliwych pozostałości (np. antybiotyki, tlenek etylenu itd.). F. OZNAKOWANIE ZEWNĘTRZNE POJEMNIKA TRANSPORTOWEGO Do celów transportowania każdy pierwotny pojemnik musi być umieszczony w pojemniku transportowym, który musi zawierać co najmniej następujące informacje: a) dane identyfikacyjne dotyczące wysyłającego banku tkanek, wraz z adresem i numerem telefonu; b) dane identyfikacyjne dotyczące organizacji odpowiedzialnej za stosowanie tkanek i komórek u ludzi, dla której przeznaczony jest pojemnik, wraz z adresem i numerem telefonu; c) oświadczenie, że paczka zawiera tkankę/komórki ludzkie z informacją: "OSTROŻNIE"; d) w przypadku gdy do przeprowadzenia przeszczepu niezbędne są komórki żywe, takie jak komórki macierzyste, gamety i embriony ludzkie, należy umieścić informację: "NIE NAPROMIENIOWYWAĆ"; e) zalecane warunki transportowania (np. trzymać w niskiej temperaturze, w pozycji stojącej itd.); f) instrukcje dotyczące bezpieczeństwa/metody schładzania (w stosownych przypadkach).
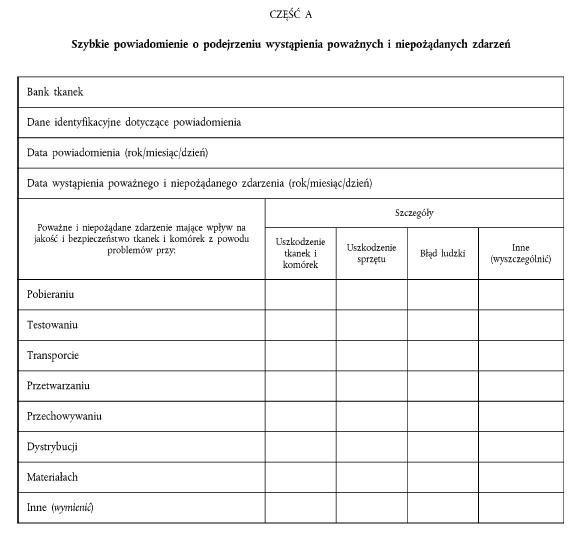
ZAŁĄCZNIK III POWIADOMIENIE O POWAŻNYCH I NIEPOŻĄDANYCH REAKCJACH
ZAŁĄCZNIK IV POWIADOMIENIE O POWAŻNYCH I NIEPOŻĄDANYCH ZDARZENIACH
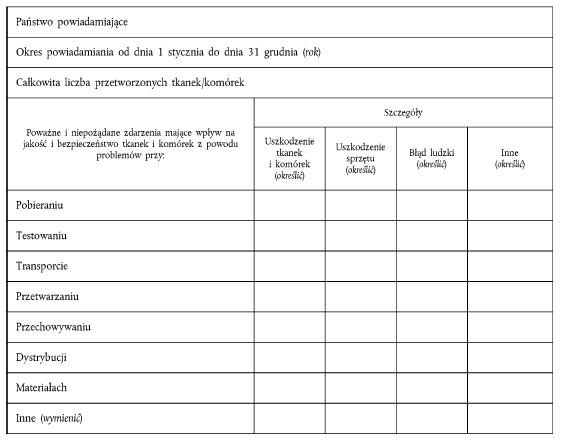
ZAŁĄCZNIK V FORMULARZ ROCZNEGO POWIADOMIENIA Część A Formularz rocznego powiadomienia o poważnych I niepożądanych reakcjach
Część B Formularz rocznego powiadomienia o poważnych I niepożądanych zdarzeniach
ZAŁĄCZNIK VI Informacje na temat minimalnych danych dawcy/biorcy, które należy zachować zgodnie z art. 9 A. DANE ZACHOWYWANE PRZEZ BANKI TKANEK Dane identyfikacyjne dawcy Dane identyfikacyjne dotyczące oddania zawierają co najmniej: - dane identyfikacyjne organizacji pobierającej lub banku tkanek, - jednolity numer identyfikacyjny oddania, - datę pobrania, - miejsce pobrania, - rodzaj oddania (np. jedna tkanka/wiele tkanek; autologiczne/alogeniczne; dawca żywy/dawca nieżywy). Dane identyfikacyjne produktu zawierają co najmniej: - dane identyfikacyjne banku tkanek, - rodzaj tkanki i komórki/produktu (nomenklatura podstawowa), - numer partii (jeśli dotyczy), - numer podgrupy (jeśli dotyczy), - data ważności, - status tkanki/komórki (tzn. poddane kwarantannie, nadające się do stosowania), - opis i pochodzenie produktów, zastosowane etapy przetwarzania, materiały i dodatki wchodzące w kontakt z tkankami i komórkami i wpływające na jakość i/lub bezpieczeństwo produktów, - dane identyfikacyjne zakładu wydającego oznaczenie końcowe. Dane identyfikacyjne dotyczące stosowania u ludzi zawierają co najmniej: - datę dystrybucji/usunięcia, - dane identyfikacyjne lekarza lub użytkownika końcowego/zakładu. B. DANE ZACHOWYWANE PRZEZ ORGANIZACJE ODPOWIEDZIALNE ZA STOSOWANIE U LUDZI a) dane identyfikacyjne banku dostarczającego tkanki; b) dane identyfikacyjne lekarza lub użytkownika końcowego/zakładu; c) rodzaj tkanek i komórek; d) dane identyfikacyjne produktu; e) dane identyfikacyjne biorcy; f) data złożenia wniosku.
ZAŁĄCZNIK VII Informacje znajdujące się w europejskim systemie kodowania a) Dane identyfikacyjne dotyczące oddania: - jednolity numer identyfikacyjny oddania, - dane identyfikacyjne banku tkanek. b) Dane identyfikacyjne produktu: - kod produktu (nomenklatura podstawowa), - numer podgrupy (jeśli dotyczy), - data ważności.
|
|
|
 Dyrektywa Komisji 2006/86/WE
Dyrektywa Komisji 2006/86/WE




Teksty aktów prawnych prezentowane na stronach internetowych Centrum Organizacyjno-Koordynacyjnego do spraw Transplantacji "Poltransplant" nie stanowią źródła prawa.
Zgodnie z Konstytucją RP oraz Ustawą z dnia 20 lipca 2000 r. o ogłaszaniu aktów normatywnych i niektórych innych aktów prawnych (Dz.U. 62, poz. 718 z późn.zm.) Jedyne źródło prawa na terenie RP stanowią akty normatywne ogłaszane w dziennikach urzędowych.
© 2024 Centrum Organizacyjno-Koordynacyjne ds. Transplantacji "Poltransplant"